목차
1. 실험제목
2. 실험목적
3. 실험 기구 및 시약
4. 실험원리
4.1 PCR
4.2 전기영동
4.3 competent cell
4.4 유전자 재조합
4.5 형질전환
4.6 Ethidium Bromide
5. 실험 방법
본문내용
1. 실험제목: 제한효소를 이용한 DNA절단& 전기 영동법 및 PCR에 의한 DNA분리 및 확인
2. 실험목적: 미생물에서 plasmid DNA를 분리하고, PCR을 이용하여 DNA를 증폭하고 제한효소를 이용하여 DNA를 절단한 후 Mini gel unit를 이용하여 전기영동법에 의해 DNA를 분리하고 확인한다.
3.실험 기구 및 시약
Gradient PCR, Mini Gel Electrophoresis Units(Mupid21), agarose gel, Microcentrifuge tube, 증류수, 제한효소, buffer용액, PCR premix tube, 증류수, primer, DMSO, 주형 DNA, size marker, circular vector plasmid, loading dye, circular recombinent plasmid
4. 실험원리
4.1 PCR
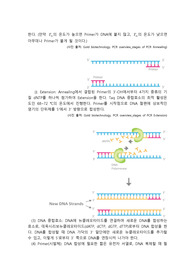
1. PCR의 원리
PCR법의 기본 원리는 다음과 같다.
첫 단계는 이중쇄 DNA를 단일쇄 DNA로 열변성(denaturation)시킨다. 두 번째 단계는 올리고 뉴클레오티드를 단일쇄 DNA에 결합(annealing)시키고 세 번째 단계는 결합한 올리고뉴클레오티드를 시발체로 하여 DNA 폴리머라제에 의해 DNA를 합성(Extension)하여 이상의 과정을 일정한 싸이클동안 반복한다.
이론적으로 매 PCR 주기마다 증폭되는 DNA는 2배씩 늘어나야 하나 (n 싸이클 후는 2n개) 여러 가지 이유에서 일정 주기가 지나면 증폭되는 DNA의 양이 안정기(plateau)에 도달하게 된다. PCR로 증폭된 밴드를 에티디움 브로마이드(EtBr)로 염색된 아가로스 젤(Agarose gel)에서 관찰하려면 약 50ng의 DNA가 필요한데 50ng은 300bp길이 DNA로 따지면 약 1011카피가 된다. 따라서 단일 copy의 300bp DNA를 증폭 후 전기영동하여 볼 수 있으려면 약 37 싸이클이면 (237=1011)된다.
출처 : 해피캠퍼스